
A large UK Biobank analysis followed 479,966 people for a median of 13.63 years and identified 6,082 new rheumatoid arthritis cases. Mobile phone users had a higher RA hazard than non-users (HR 1.14, 95% CI 1.07–1.23), and people using a phone for more than 30 minutes/week had elevated risk (HR 1.08, 95% CI 1.02–1.15). Higher weekly call-time groups also showed elevated hazards, while hands-free/speakerphone use was not statistically significant.
This does not by itself prove causation. Phone use is a composite exposure: RF-EMF, near-field pulsing, behavior, blue light/circadian disruption, and sedentary time can travel together. The authors themselves note unclear mechanisms and residual confounding, and they call for future causal work.
But the cleanest biological theory is this:
Bioelectric Timing Fidelity Loss.
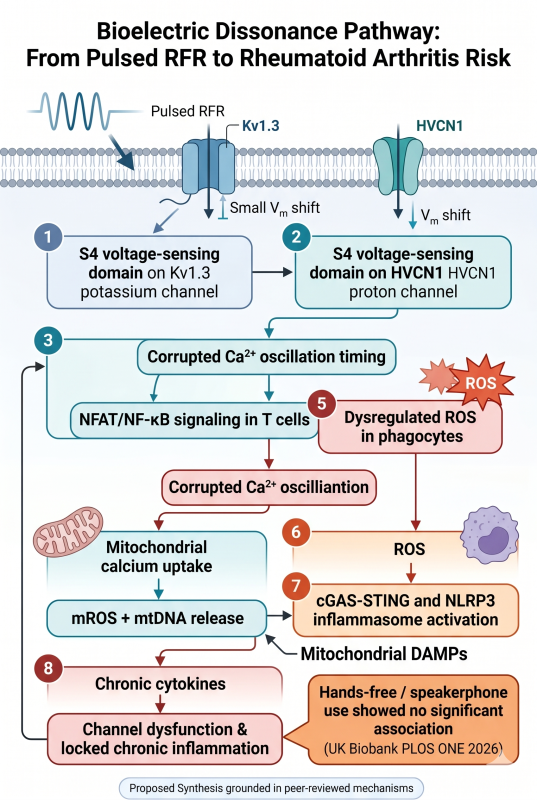
Immune cells are not just chemical systems. They are electrically timed decision systems. Voltage-gated ion channels use S4 voltage-sensor segments to convert membrane-potential changes into channel opening and closing; S4 movement is a core electromechanical switch in voltage-gated channels.
T cells decode antigenic context through calcium timing, potassium-channel support, and transcriptional interpretation. Kv1.3 and KCa3.1 help maintain the electrochemical driving force for sustained calcium entry, and calcium influx is described as a cornerstone of T-lymphocyte activation and function. NFAT, one of the major immune transcriptional decoders, is controlled by calcium influx downstream of T-cell receptor signaling.
So the proposed pathway is:
time-structured EMF exposure → membrane/electrical timing stress → altered S4/channel gating probability or ion-flux timing → distorted Ca²⁺/H⁺/K⁺ signaling → NFAT/NF-κB/AP-1 misinterpretation → oxidative stress → mitochondrial DAMP release → innate immune activation → loss of tolerance → autoimmune-like escalation.
The central claim is not that RF “heats tissue until disease happens.” The stronger claim is that immune signaling depends on timing fidelity, and a chronic, pulsed environmental signal could act as biological timing noise in susceptible people.
The redox side of the model is also biologically coherent. Hv1/HVCN1 proton channels are required for high-level NADPH-oxidase-dependent superoxide production during phagocyte respiratory burst, and HVCN1 also modulates B-cell receptor signaling, mitochondrial metabolism, and antibody responses.
Once calcium/proton/redox timing is distorted, mitochondria become the amplifier. Oxidative stress and mitochondrial dysfunction can release mitochondrial DNA into the cytosol or extracellular space, where mtDNA functions as a DAMP and activates innate immune pathways such as cGAS-STING, TLR9, and inflammasome signaling, producing type I interferons and pro-inflammatory cytokines.
That is exactly the kind of biology that can feed RA. RA pathogenesis involves modified self-antigens, autoantibodies such as RF and ACPA, immune complexes, macrophage activation, TNF, IL-6, IL-17, synovial fibroblast activation, and osteoclast-mediated tissue destruction.
The honest scientific position is this: the downstream immune biology is strong; the epidemiologic signal is now visible; the RF-to-S4/ion-timing bridge remains the key contested step. A skeptical ARPANSA/Radiation Research review concluded that RF-induced currents at ICNIRP guideline limits are many orders of magnitude below those needed to affect VGCC gating and that experimental studies have not validated RF effects on calcium transport.
At the same time, oxidative-stress evidence remains biologically suggestive but heterogeneous. Schuermann and Mevissen reported that many animal and cell studies show increased oxidative stress after RF-EMF or ELF-MF exposure, while a 2024 systematic review judged the overall certainty for RF-EMF effects on oxidative-stress biomarkers to be very low because of inconsistency, heterogeneity, and risk of bias.
So the best theory is not “RF directly causes RA in everyone.”
The best theory is:
In genetically or immunologically susceptible people, chronic pulsed phone-related exposure may act as a low-grade bioelectric timing stressor. If it degrades ion-channel timing fidelity in immune cells, that can shift calcium/redox signaling toward sterile danger, mitochondrial DAMP release, inflammatory cytokines, and loss of self-tolerance. RA would be one possible endpoint when this occurs in a host already primed for citrullinated-antigen autoimmunity.
The NTP animal data should be used carefully. NTP found that high RFR exposure was associated with clear evidence of malignant heart schwannomas in male rats and significant DNA damage in selected tissues, but it did not test RA or prove an autoimmune S4 pathway. It is best cited only as evidence that sufficiently intense RFR can produce biological effects in specific tissues under controlled animal-exposure conditions.
Bottom line: this PLOS ONE study does not close the case. It opens the right mechanistic case:
mobile-phone exposure → bioelectric timing disruption → immune ion-flux mis-coding → oxidative/DAMP signaling → tolerance failure → autoimmune risk.
That is a falsifiable theory. Now it needs decisive experiments.
The pathway, step by step
1. S4 is the voltage-sensing gate.
Voltage-gated channels use S4 gating charges to translate membrane electrical state into pore opening/closing. This part is established channel biophysics.
2. Immune cells depend on ion timing.
T-cell activation depends on calcium influx, supported by Kv1.3/KCa3.1 potassium-channel control of membrane potential; NFAT is a calcium-controlled transcriptional decoder of T-cell activation and differentiation.
3. The RF/ELF-to-channel bridge is the hypothesis, not settled proof.
This is the vulnerable link. A world-class theory must admit it. The claim must be tested as timing perturbation, not assumed as “RF opens channels.”
4. Ion timing errors can become redox errors.
Hv1/HVCN1 couples proton movement to NADPH oxidase superoxide production in phagocytes and affects B-cell receptor signaling/metabolism. That gives a direct immune-channel-to-ROS bridge.
5. Redox stress can become sterile danger signaling.
Mitochondrial stress and mtDNA release can activate cGAS-STING, TLR9, inflammasome pathways, type I interferons, and inflammatory cytokines.
6. Sterile danger plus modified self-antigen can become RA biology.
In RA, ACPA/RF immune complexes, macrophage activation, TNF, IL-6, IL-17, synovial fibroblast activation, and osteoclastogenesis form a self-reinforcing inflammatory loop.
The decisive experiment
To prove this pathway, the field needs a blinded, temperature-controlled, waveform-specific study using primary human immune cells:
Expose naïve T cells, memory T cells, B cells, monocytes/macrophages, and dendritic cells to realistic pulsed RF/ELF-modulated signals and sham controls. Measure, in the same experiment, membrane potential, Kv1.3/KCa3.1/CRAC/Hv1 currents, Ca²⁺ oscillation frequency, NFAT/NF-κB nuclear translocation, mitochondrial ROS, cytosolic mtDNA, cGAS-STING/TLR9/NLRP3 activation, IL-1β, IL-6, TNF-α, type I IFN, Th17/Treg skewing, and antigen-presentation markers.
Then add rescue tests: Kv1.3 blockers, CRAC/Orai blockers, Hv1 inhibition, mitochondrial ROS scavengers, STING/TLR9/NLRP3 blockade, and genetic/channel perturbations. If exposure effects disappear when the ion-channel/redox/DAMP nodes are blocked, the pathway becomes much stronger.
That is the “proof chain” to demand:
RF waveform specificity → S4/ion-channel timing shift → calcium/proton/redox disruption → mitochondrial DAMP release → innate immune activation → adaptive tolerance failure.






